-
Address
209, Veritas A Hall, Yonsei Univeristy, 85 Songdogwahak-ro, Yeonsu-gu, Incheon 21983, Republic of Korea -
Phone
032-212-9550 -
FAX
032-212-9572 -
E-mail
support@bmdrc.org



Computer-Aided Drug Design

Computer-Aided Drug Design

Computer-Aided Drug Design
HOME > Services & Products > Computer-Aided Drug Design
Target Identification & Validation
for Small Molecule Drug Discovery
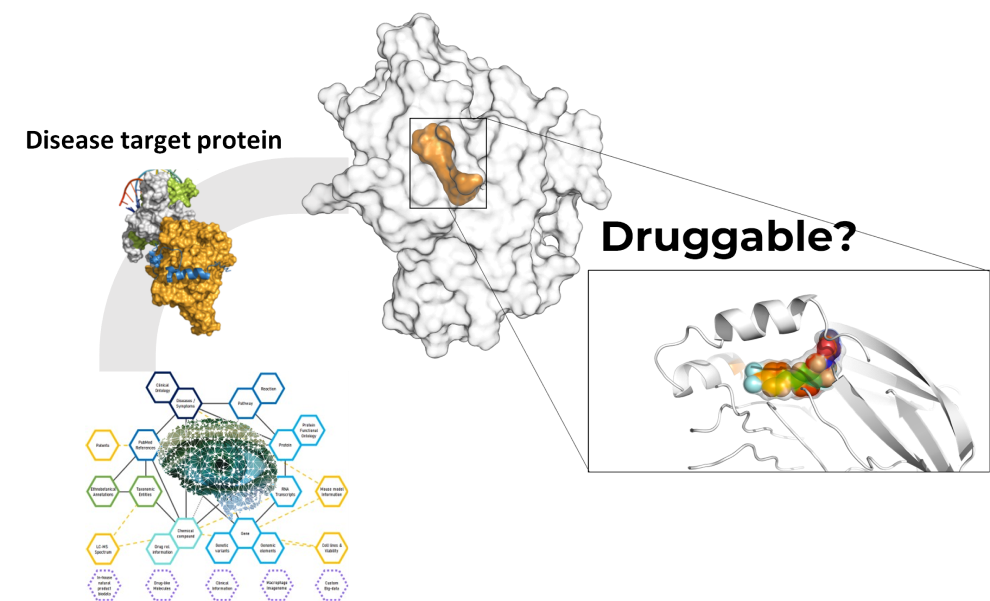
Suggestion & Validation of Druggable Target
for Customer Interest
- We utilize an AI network platform to provide appropriate target proteins for diseases that customers are interested in. This is an important task in the field of drug development, as it can help develop new treatments.
- The AI network platform helps analyze large amounts of data to identify suitable target proteins. This can help discover new treatments for diseases that currently have limited treatment options.
- We can evaluate the druggability of the proposed disease-related target protein using structure-based molecular modeling to analyze the protein's structure, active site, and surrounding regions. This approach enables us to assess the protein's druggability and identify potential small molecules for further drug discovery. The assessment of the target protein's druggability and identification of suitable compounds for further development is a critical step in the drug discovery process.
Small Molecule Discovery
for Target Protein
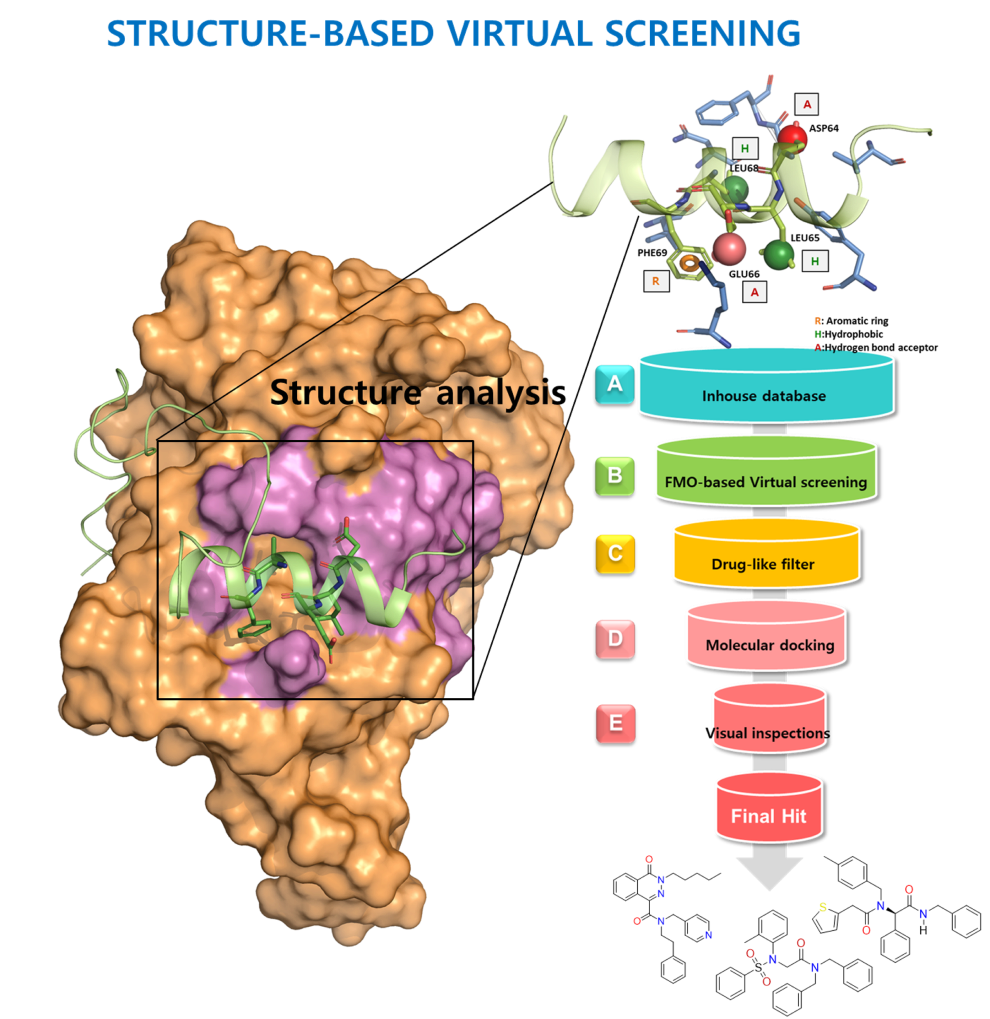
Small Molecule Discovery for Target Protein
by Virtual Screening
- Module for PK predictionProtein structure-based virtual screening is a technique that uses computer simulations to identify small molecules that interact with a protein's structure. This technique can be used to understand the mechanism of action of a particular protein and identify small molecule inhibitors, which can inhibit its function..
- The structure of the target protein is first determined and then simulated using computer modeling. A large database of compounds is then searched for compounds that are likely to interact with the protein. Molecular dynamics simulations, artificial intelligence, and machine learning are often used in this process.
- The virtual hit molecules identified through virtual screening are subjected to various experimental validations, and ultimately a small molecule inhibitor with high potential is selected.
Hit Compound Structure Optimization
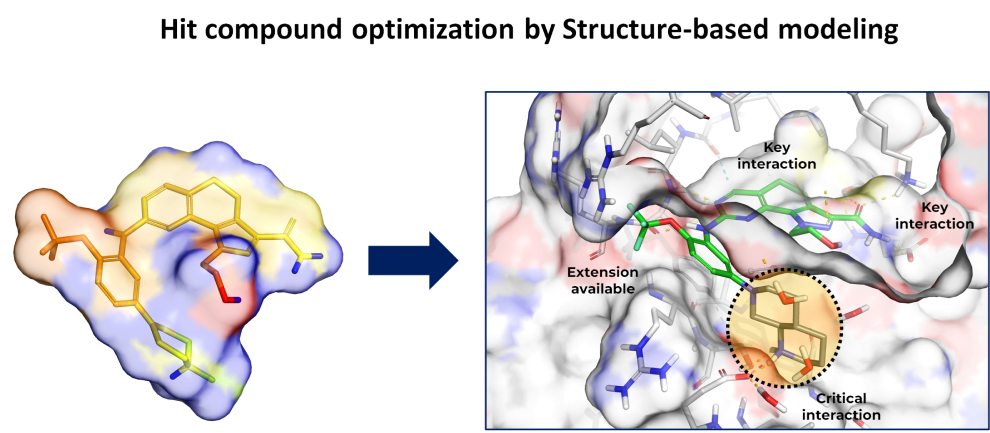
Hit Compound Structure Optmization
by Computational Method
- Hit structure optimization is a critical step in hit-to-lead optimization of virtual screening compounds by structure-based modeling. The goal of this step is to improve the potency and selectivity of the initial hit compound identified by virtual screening through rational design and optimization of its structure using computational methods.
- Hit structure optimization by structure-based modeling is a highly iterative process that requires expertise in both computational and medicinal chemistry. However, when done effectively, it can significantly accelerate the drug discovery process and lead to the identification of novel and effective therapies for a wide range of diseases.
Hit-to-Lead Optimization
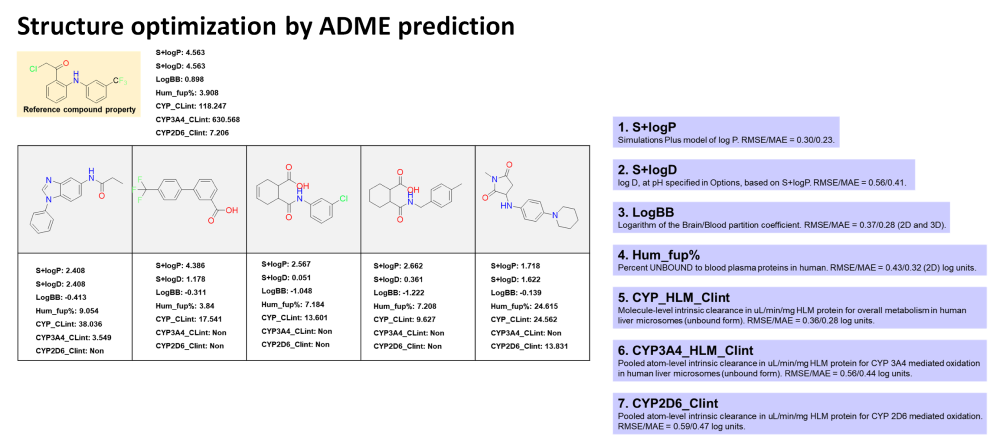
Hit-to-Lead Optimization by Predicting
ADME Properties
- Lead optimization by computational methods involves the use of various computational approaches to refine the chemical structure of a lead compound, improving its potency, selectivity, and other properties, while reducing its toxicity and increasing its pharmacokinetic properties.
- Accurately predicting the absorption, distribution, metabolism, excretion (ADME), and toxicity of the lead compounds is an essential part of the optimization process.
Ligand-based Drug-Design
Focuses on Designing New Drugs based on the Properties and Interactions of Given Ligands
- A library of small molecules (ligands) is first screened to identify molecules that are likely to bind to the target protein of interest. Next, the identified ligands are optimized through chemical modification to enhance their binding affinity and selectivity towards the target protein.
- The design process often involves the use of computer-aided techniques, such as molecular docking and quantitative structure-activity relationship (QSAR) modeling, to predict the binding interactions between the ligands and the target protein, as well as to optimize the properties of the ligands.
- Ligand-based drug design can be a powerful tool in the development of new drugs, particularly when the 3D structure of the target protein is known, as it allows for the rational design of compounds with improved potency and selectivity.
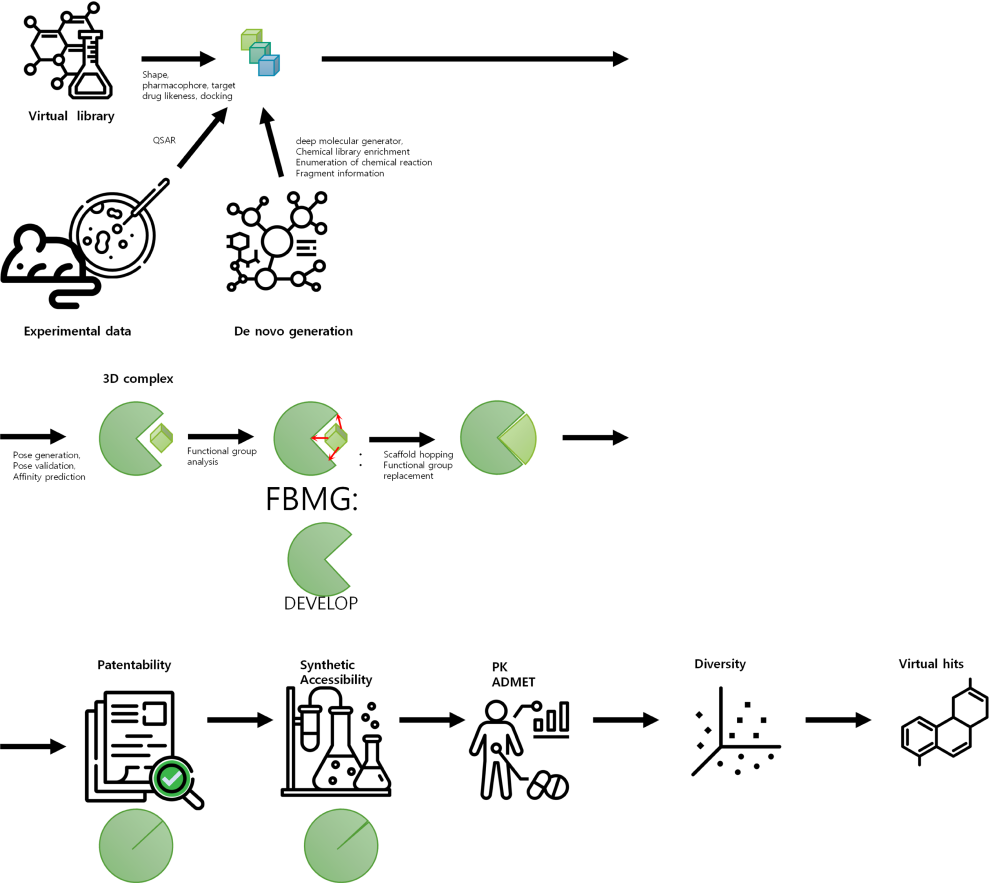
Related Research Fields





CONTACT US
Contact Us